What Is Adipotide? The Prohibitin-Targeting Peptide Studied for Fat Vasculature
Adipotide (FTPP / Prohibitin-TP01) is a chimeric peptidomimetic studied for targeting white-fat blood vessels. What the primate and mouse data actually show — RUO.
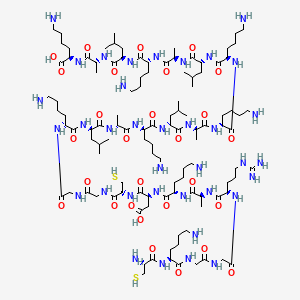
Adipotide is a synthetic chimeric peptidomimetic, sequence CKGGRAKDC-GG-D(KLAKLAK)2, studied in mice and obese monkeys. Its homing domain binds prohibitin on white-fat blood vessels; the attached proapoptotic motif was reported to ablate that vasculature. It was never approved; human development was discontinued in 2019.
Adipotide is one of the more mechanistically unusual anti-obesity candidates ever put into animals: rather than acting on appetite, hormones, or fat cells, it was engineered to attack the blood supply of white adipose tissue. It carries two halves — a peptide that homes to a protein called prohibitin on fat-feeding vessels, and a second peptide that kills the cell it lands on. Reported results in obese monkeys drew a lot of attention, but the compound never cleared human testing. Everything below describes laboratory and literature findings in mice and non-human primates, not use in people.
What is adipotide, structurally?
Adipotide goes by several names — FTPP (Fat-Targeted Proapoptotic Peptide), Prohibitin-Targeting Peptide 1, Prohibitin-TP01 — and is best identified not by a trade name but by its published sequence: CKGGRAKDC-GG-D(KLAKLAK)2.1 It is a chimera of two functional modules. The first is CKGGRAKDC, a nine-residue cyclic homing peptide closed by a disulfide bond between Cys1 and Cys9. The second, joined through a two-glycine linker, is D(KLAKLAK)2 — a fourteen-residue amphipathic sequence built from D-amino acids. The D-stereochemistry matters: it makes the motif resistant to the proteases that would chew up a normal L-peptide, and this fragment is a known membrane-disrupting, proapoptotic element that damages mitochondria once it gets inside a cell.
By molecular weight the assembled molecule sits around 2,557–2,611 Da across 25 residues (nine in the homing domain, two glycines, fourteen in the proapoptotic tail). A CAS number of 859216-15-2 is commonly cited via PubChem and Wikipedia,8 but that identifier is not canonical across suppliers — some list 1422956-49-3 instead. For a disulfide-cyclised, D-amino-acid chimera like this, the sequence plus a batch certificate of analysis is a far more reliable identity handle than a CAS lookup.
How was it designed to work?
The logic behind adipotide came out of a simple observation about fat: like a tumour, an expanding adipose depot needs a blood supply, and that vasculature is potentially a target. The foundational work was the 2004 mouse study by Kolonin and colleagues, which used in vivo phage display — screening enormous combinatorial peptide libraries in a living animal — to fish out short peptides that home specifically to the blood vessels of white fat.2 That screen produced the CKGGRAKDC sequence and identified its receptor as prohibitin, a mitochondrial and membrane protein that turns out to be displayed on the endothelium of fat-feeding vessels. Fused to a proapoptotic cargo, the peptide ablated white adipose tissue and reversed obesity in the mice.
The assembled molecule therefore has a two-step logic: the CKGGRAKDC domain acts as a postal code, docking onto prohibitin (and, in later human-tissue mapping, annexin A2) on white-fat endothelium; the D(KLAKLAK)2 domain then acts as the payload, triggering apoptosis of that endothelial cell.4 The consequence, as reported, is that vessels feeding the fat depot regress and the depot shrinks for lack of perfusion. The point worth holding onto is where it acts — on the vasculature, not on adipocytes directly and not on any central appetite circuit. That conceptual foundation was not unique to this group: the Folkman lab had already shown in 2002 that anti-angiogenic agents caused reversible adipose loss in obese mice, establishing that fat mass is genuinely vasculature-dependent.5
4 The total number of participants ever enrolled in the only human trial of adipotide before it was terminated.
What the primate study reported
The paper that made adipotide well known is Barnhart et al., published in Science Translational Medicine in 2011.1 In spontaneously obese Old World monkeys, a 28-day treatment cycle was associated with fat loss and improved insulin resistance, with the reduction in white adipose tissue documented by MRI and DXA imaging rather than the scale alone. Secondary summaries frequently attach a figure of roughly 11% body-weight reduction over about four weeks, but it is worth being precise: the peer-reviewed abstract describes “rapid weight loss” and does not foreground a single headline percentage. The ~11% number circulates mainly through secondary sources, so it should be quoted with that caveat.
The study reported fat loss in obese monkeys, and in the same breath reported changes in kidney function — the toxicity was never a hidden appendix.
| Stage | Model | What was reported |
|---|---|---|
| Target discovery (2004) | Mice | CKGGRAKDC homes to white-fat vasculature; prohibitin identified as receptor; obesity reversal2 |
| Vascular receptor mapping (2011) | Human tissue screen | Prohibitin + annexin A2 confirmed on human white-fat vasculature4 |
| Lead efficacy study (2011) | Obese monkeys | Fat loss, improved insulin resistance; reversible renal proximal-tubule changes1 |
| Human trial (2012–2019) | Phase I, n=4 | Terminated; no efficacy/safety data published; discontinued6 |
All rows describe literature and in-vivo animal findings, or an incomplete human trial with no published outcomes. None of this represents a use recommendation, dosing scheme, or safety assessment for any organism.
An honest read of the evidence
This is a compound where the honest read is more important than the headline, and there are several reasons to be cautious rather than impressed.
First, renal toxicity is not buried — it is stated in the abstract of the lead paper itself. Barnhart et al. report dose-dependent changes in renal proximal tubule function in monkeys of three species.1 The authors describe these changes as reversible, but dose-limiting effects on the kidney are widely understood to be the reason human development stalled. Any account of adipotide that leads with the fat-loss number and treats the kidney signal as a footnote has the emphasis backwards.
Second, there is no successful human clinical development to point to. The only trial in people — a first-in-man Phase I study in obese men with metastatic prostate cancer, NCT01262664 — was terminated after enrolling just four subjects.6 No peer-reviewed human efficacy or full safety data were ever published. Development was formally discontinued in January 2019, when Ablaris Therapeutics ended the trial at the principal investigator’s request; the compound was never approved and no EMA application was filed.7 The trial record itself states the material “is not FDA approved or commercially available.”
Third, essentially all of the primary data trace to a single research lineage — the Arap, Pasqualini and Kolonin group at MD Anderson — commercialised through Ablaris/Alvos and Arrowhead Therapeutics. Members of that group held equity in the developing companies, disclosed in the 2011 human-tissue mapping paper.4 That is not an accusation of misconduct; it is a reason the absence of independent replication matters. No outside laboratory has reproduced the primate fat-loss result.
Fourth, the mechanism itself is contested. A published Comment by Criscione in Science Translational Medicine argued that the observed fat loss in the monkeys might reflect a direct effect on food intake rather than the claimed selective vascular apoptosis.3 Whether the headline “attacks fat’s blood supply” mechanism is even the operative one is, on the published record, unsettled. Layered on top is the ordinary species-translation problem: efficacy was shown only in rodents and non-human primates, the primate numbers lean on secondary summaries, and anti-obesity agents have a poor historical record of surviving the jump from primates to humans — a point the paper’s own authors acknowledge.
Related research on other metabolic and exercise-linked compounds sits in the same “interesting mechanism, immature evidence” category — see our notes on AOD-9604, tesofensine, and the broader landscape of exercise mimetics.
All materials supplied by Condor Research are Research Use Only (RUO). Everything above describes in-vitro, animal, and literature findings — not a dosing protocol, clinical guidance, or safety assessment for any organism, human or veterinary. Adipotide is an investigational compound with no approved use anywhere.
Condor Research · Sportello scientifico
Atrio Sciences s.r.o., IČO 57 669 651, Nitra (SK) · info@condorresearch.com
- Adipotide (FTPP; Prohibitin-Targeting Peptide 1) is a chimeric peptidomimetic: a cyclic prohibitin-homing domain (CKGGRAKDC, Cys1-Cys9 disulfide) joined by a Gly-Gly linker to a D-amino-acid proapoptotic motif, D(KLAKLAK)2.
- It was designed to act on the vasculature feeding white adipose tissue — the homing domain binds prohibitin (and annexin A2) on those endothelial cells — not on fat cells directly and not on the brain.
- The lead primate study (Barnhart et al., Sci Transl Med 2011) reported fat loss and improved insulin resistance in obese Old World monkeys, confirmed by MRI and DXA.
- The same abstract reports dose-dependent, described-as-reversible changes in renal proximal tubule function — an in-abstract toxicity signal, not a footnote.
- Human development failed: the only clinical trial (Phase I, NCT01262664) was terminated after 4 participants; development was formally discontinued in January 2019, with no approval anywhere.
- The entire primary evidence base comes from a single research lineage (the Arap/Pasqualini/Kolonin group at MD Anderson), with no independent replication of the primate result and a published mechanistic critique.
Is adipotide the same as a GLP-1 drug?
No. It is mechanistically unrelated. GLP-1 receptor agonists act on incretin signalling and appetite, whereas adipotide was designed to home to prohibitin on white-fat blood vessels and trigger apoptosis there. The two share nothing in structure or target.
Why did human development stop?
The single Phase I trial (NCT01262664) was terminated after only four participants, and development was formally discontinued in January 2019. Dose-limiting effects on renal proximal tubule function reported in the primate study are widely understood to be the underlying obstacle.
What does prohibitin have to do with fat?
Prohibitin is a protein displayed on the endothelium of blood vessels that supply white adipose tissue. In vivo phage-display screening identified it as the receptor for the CKGGRAKDC homing peptide, making it a vascular "address" for that fat depot. Later work mapped prohibitin, together with annexin A2, on human white-fat vasculature as well.
Is the ~11% weight-loss figure reliable?
Treat it carefully. That percentage appears mainly in secondary summaries; the peer-reviewed abstract describes "rapid weight loss" in obese monkeys without foregrounding a single headline number. The imaging data (MRI, DXA) support a marked reduction in white adipose tissue, but the specific percentage should not be quoted as if it came straight from the primary paper.
Is the vascular-apoptosis mechanism settled?
No. A published Comment argued the fat loss might instead reflect a direct effect on food intake rather than the claimed selective ablation of fat vasculature. The mechanism as usually described is plausible and grounded in the homing/receptor data, but it has been formally challenged in the literature and is not considered closed.
How should adipotide's identity be verified?
By its published sequence, CKGGRAKDC-GG-D(KLAKLAK)2, plus a batch certificate of analysis — not by CAS number. Multiple CAS numbers circulate (859216-15-2 and 1422956-49-3 among them), and for a disulfide-cyclised, D-amino-acid chimera the sequence and analytical data are the dependable identifiers.